{kind=link}
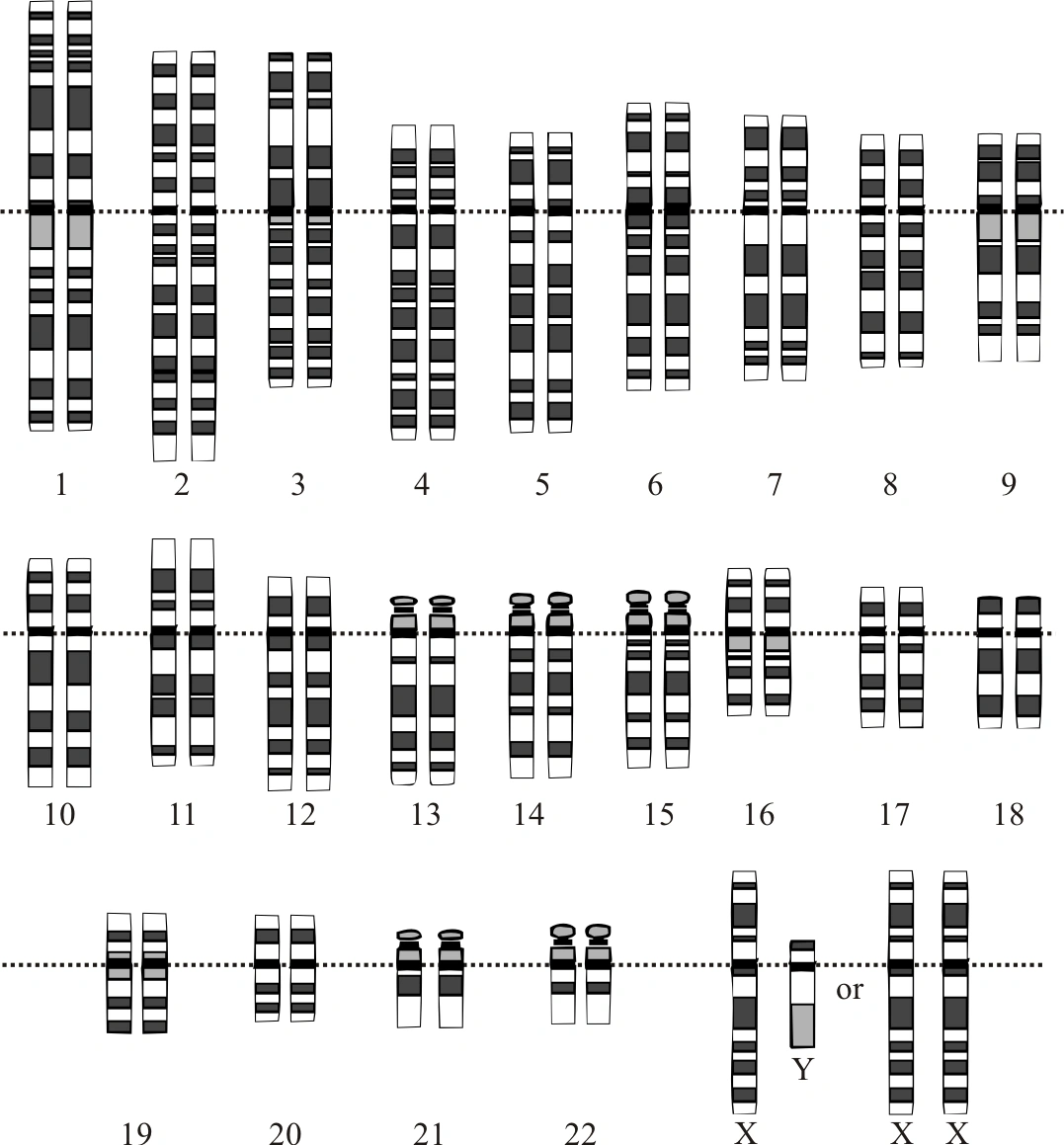
Graphical representation of the idealized human diploid karyotype, showing the organization of the genome into chromosomes. This drawing shows both the female (XX) and male (XY) versions of the 23rd chromosome pair. Chromosomes are shown aligned at their centromeres. The mitochondrial DNA is not shown.
The human (Homo sapiens) genome is the complete set of human genetic information, stored as DNA sequences within the 23 chromosome pairs of the cell nucleus and in a small DNA molecule within the mitochondrion. The haploid human genome (contained in egg and sperm cells) consists of three billion DNA base pairs, while the diploid genome (found in somatic cells) has twice the DNA content. Although the human genome has been completely read by DNA sequencing, it is not yet fully understood. Many (though probably not all) genes have been identified, yet much work still needs to be done to completely elucidate the biological functions of their protein and RNA products. Recent results indicate that most of the vast quantities of noncoding DNA within the genome have associated biochemical activities, including regulation of gene expression, organization of chromosome architecture, and signals controlling epigenetic inheritance.
The Human Genome Project produced the first complete sequences of individual human genomes. As of 2012, thousands of human genomes have been completely sequenced, and many more have been mapped at lower levels of resolution. The resulting data are used worldwide in biomedical science, anthropology, forensics and other branches of science. There is a widely-held expectation that genomic studies will lead to advances in the diagnosis and treatment of diseases, and to new insights in many fields of biology, including human evolution.
The haploid human genome contains approximately 20,000 protein-coding genes, significantly fewer than had been anticipated.[1][2] Protein-coding sequences account for only a very small fraction of the genome (approximately 1.5%), and the rest is associated with non-coding RNA molecules, regulatory DNA sequences, introns, and sequences to which no function has yet been assigned.[3]

{kind=link}
This figure illustrates the proportion of various genomic components of the human genome.
Molecular organization and gene content[]
The human genome is organized into chromosomes (very large linear DNA molecules contained within the cell nucleus) and the small mitochondrial DNA (a comparatively tiny circular DNA molecule). Basic information about these molecules and their gene contents are provided in the following table. (Data source: Ensembl genome browser release 68, July 2012)
| Chromosome | Length (mm) | Base pairs | Variations | Confirmed proteins | Putative proteins | Pseudogenes | miRNA | rRNA | snRNA | snoRNA | Misc ncRNA | Links |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 85 | 249,250,621 | 4,401,091 | 2,012 | 31 | 1,130 | 134 | 66 | 221 | 145 | 106 | EBI |
| 2 | 83 | 243,199,373 | 4,607,702 | 1,203 | 50 | 948 | 115 | 40 | 161 | 117 | 93 | EBI |
| 3 | 67 | 198,022,430 | 3,894,345 | 1,040 | 25 | 719 | 99 | 29 | 138 | 87 | 77 | EBI |
| 4 | 65 | 191,154,276 | 3,673,892 | 718 | 39 | 698 | 92 | 24 | 120 | 56 | 71 | EBI |
| 5 | 62 | 180,915,260 | 3,436,667 | 849 | 24 | 676 | 83 | 25 | 106 | 61 | 68 | EBI |
| 6 | 58 | 171,115,067 | 3,360,890 | 1,002 | 39 | 731 | 81 | 26 | 111 | 73 | 67 | EBI |
| 7 | 54 | 159,138,663 | 3,045,992 | 866 | 34 | 803 | 90 | 24 | 90 | 76 | 70 | EBI |
| 8 | 50 | 146,364,022 | 2,890,692 | 659 | 39 | 568 | 80 | 28 | 86 | 52 | 42 | EBI |
| 9 | 48 | 141,213,431 | 2,581,827 | 785 | 15 | 714 | 69 | 19 | 66 | 51 | 55 | EBI |
| 10 | 46 | 135,534,747 | 2,609,802 | 745 | 18 | 500 | 64 | 32 | 87 | 56 | 56 | EBI |
| 11 | 46 | 135,006,516 | 2,607,254 | 1,258 | 48 | 775 | 63 | 24 | 74 | 76 | 53 | EBI |
| 12 | 45 | 133,851,895 | 2,482,194 | 1,003 | 47 | 582 | 72 | 27 | 106 | 62 | 69 | EBI |
| 13 | 39 | 115,169,878 | 1,814,242 | 318 | 8 | 323 | 42 | 16 | 45 | 34 | 36 | EBI |
| 14 | 36 | 107,349,540 | 1,712,799 | 601 | 50 | 472 | 92 | 10 | 65 | 97 | 46 | EBI |
| 15 | 35 | 102,531,392 | 1,577,346 | 562 | 43 | 473 | 78 | 13 | 63 | 136 | 39 | EBI |
| 16 | 31 | 90,354,753 | 1,747,136 | 805 | 65 | 429 | 52 | 32 | 53 | 58 | 34 | EBI |
| 17 | 28 | 81,195,210 | 1,491,841 | 1,158 | 44 | 300 | 61 | 15 | 80 | 71 | 46 | EBI |
| 18 | 27 | 78,077,248 | 1,448,602 | 268 | 20 | 59 | 32 | 13 | 51 | 36 | 25 | EBI |
| 19 | 20 | 59,128,983 | 1,171,356 | 1,399 | 26 | 181 | 110 | 13 | 29 | 31 | 15 | EBI |
| 20 | 21 | 63,025,520 | 1,206,753 | 533 | 13 | 213 | 57 | 15 | 46 | 37 | 34 | EBI |
| 21 | 16 | 48,129,895 | 787,784 | 225 | 8 | 150 | 16 | 5 | 21 | 19 | 8 | EBI |
| 22 | 17 | 51,304,566 | 745,778 | 431 | 21 | 308 | 31 | 5 | 23 | 23 | 23 | EBI |
| X | 53 | 155,270,560 | 2,174,952 | 815 | 23 | 780 | 128 | 22 | 85 | 64 | 52 | EBI |
| Y | 20 | 59,373,566 | 286,812 | 45 | 8 | 327 | 15 | 7 | 17 | 3 | 2 | EBI |
| mtDNA | 0.0054 | 16,569 | 929 | 13 | 0 | 0 | 0 | 2 | 0 | 0 | 22 | EBI |
Table 1 (above) summarizes the physical organization and gene content of the human reference genome, with links to the original analysis, as published in the Ensembl database at the European Bioinformatics Institute (EBI) and Wellcome Trust Sanger Institute. Chromosome lengths were estimated by multiplying the number of base pairs by 0.34 nanometers, the distance between base pairs in the DNA double helix. The number of proteins is based on the number of initial precursor mRNA transcripts, and does not include products of alternative pre-mRNA splicing, or modifications to protein structure that occur after translation. The number of variations is a summary of unique DNA sequence changes that have been identified within the sequences analyzed by Ensembl as of July, 2012; that number is expected to increase as further personal genomes are sequenced and examined. In addition to the gene content shown in this table, a large number of non-expressed functional sequences have been identified throughout the human genome (see below). Links open windows to the reference chromosome sequence in the EBI (E), NCBI (N), or UCSC (U) genome browsers. The table also describes prevalence of genes encoding structural RNAs in the genome. MiRNA, or MicroRNA, functions as a post-transcriptional regulator of gene expression. Ribosomal RNA, or rRNA, makes up the RNA portion of the ribosome and is critical in the synthesis of proteins. Small nuclear RNA, or snRNA, is found in the nucleus of the cell. Its primary function is in the processing of pre-mRNA molecules and also in the regulation of transcription factors. SnoRNA, or Small nucleolar RNA, primarily functions in guiding chemical modifications to other RNA molecules.
Coding vs. noncoding DNA[]
The content of the human genome is commonly divided into coding and noncoding DNA sequences. Coding DNA is defined as those sequences that can be transcribed into mRNA and translated into proteins during the human life cycle; these sequences occupy only a small fraction of the genome (<2%). Noncoding DNA is made up of all of those sequences (ca. 98% of the genome) that are not used to encode proteins. Some noncoding DNA contains genes for RNA molecules with important biological functions (noncoding RNA, for example ribosomal RNA and transfer RNA). The exploration of the function and evolutionary origin of noncoding DNA is an important goal of contemporary genome research, including the ENCODE (Encyclopedia of DNA Elements) project, which aims to survey the entire human genome, using a variety of experimental tools whose results are indicative of molecular activity.
Coding sequences (protein-coding genes)[]

{kind=link}
Human proteins categorized by function, given both as number of encoding genes and percentage of all genes.[4]
Protein-coding sequences represent the most widely studied and best understood component of the human genome. These sequences ultimately lead to the production of all human proteins, although several biological processes (e.g. DNA rearrangements and alternative pre-mRNA splicing) can lead to the production of many more unique proteins than the number of protein-coding genes. The complete modular protein-coding capacity of the genome is contained within the exome, and consists of DNA sequences encoded by exons that can be translated into proteins. Because of its biological importance, and the fact that it constitutes less than 2% of the genome, sequencing of the exome was the first major milepost of the Human Genome Project.
The number of protein-coding genes within the human genome remains a subject of active investigation. A 2012 analysis of the human genome based on in vitro gene expression in multiple cell lines identified 20,687 protein-coding genes.[2] Historically, the estimate of the number of protein genes has varied widely, from as many as 2,000,000 in the late 1960s [5] to approximately 40,000.[6] Remarkably, the number of human protein-coding genes is significantly smaller than that of many less complex organisms, such as the roundworm and the fruit fly. This difference may result from the extensive use of alternative pre-mRNA splicing in humans, which provides the ability to build a very large number of modular proteins through the selective incorporation of exons
Protein-coding genes are distributed unevenly across the chromosomes, with an especially high gene density within chromosomes 19, 11, and 1 (Table 1). Each chromosome contains various gene-rich and gene-poor regions, which may be correlated with chromosome bands and GC-content . The significance of these nonrandom patterns of gene density is not well understood.[7]
The size of protein-coding genes within the human genome shows enormous variability (Table 2). For example, the gene for histone H1a (HIST1HIA) is relatively small and simple, lacking introns and encoding mRNA sequences of 781 nt and a 215 amino acid protein (648 nt open reading frame). Dystrophin (DMD) is the largest protein-coding gene in the human reference genome, spanning a total of 2.2 MB, while Titin (TTN) has the longest coding sequence (80,780 bp), the largest number of exons (364), and the longest single exon (17,106 bp). Over the whole genome, the median size of an exon is 122 bp (mean = 145 bp), the median number of exons is 7 (mean = 8.8), and the median coding sequence encodes 367 amino acids (mean = 447 amino acids; Table 21 in [3]).
| Protein | Chrom | Gene | Length | Exons | Exon length | Intron length | Alt splicing |
|---|---|---|---|---|---|---|---|
| Breast cancer type 2 susceptibility protein | 13 | BRCA2 | 83,736 | 27 | 11,386 | 72,350 | yes |
| Cystic fibrosis transmembrane conductance regulator | 7 | CFTR | 202,881 | 27 | 4,440 | 198,441 | yes |
| Cytochrome b | MT | MTCYB | 1,140 | 1 | 1,140 | 0 | no |
| Dystrophin | X | DMD | 2,220,381 | 79 | 10,500 | 2,209,881 | yes |
| Glyceraldehyde-3-phosphate dehydrogenase | 12 | GAPDH | 4,444 | 9 | 1,425 | 3,019 | yes |
| Hemoglobin beta subunit | 11 | HBB | 1,605 | 3 | 626 | 979 | no |
| Histone H1A | 6 | HIST1H1A | 781 | 1 | 781 | 0 | no |
| Titin | 2 | TTN | 281,434 | 364 | 104,301 | 177,133 | yes |
Table 2. Examples of human protein-coding genes. Chrom, chromosome. Alt splicing, alternative pre-mRNA splicing. (Data source: Ensembl genome browser release 68, July 2012)
Noncoding DNA (ncDNA)[]
Noncoding DNA is defined as all of the DNA sequences within a genome that are not found within protein-coding exons, and so are never represented within the amino acid sequence of expressed proteins. By this definition, more than 98% of the human genomes is comprised of ncDNA.
Numerous classes of noncoding DNA have been identified, including genes for noncoding RNA (e.g. tRNA and rRNA), pseudogenes, introns, untranslated regions of mRNA, regulatory DNA sequences, repetitive DNA sequences, and sequences related to mobile genetic elements.
Numerous sequences that are included within genes are also defined as noncoding DNA. These include genes for noncoding RNA (e.g. tRNA, rRNA), and untranslated components of components of protein-coding genes (e.g. introns, and 5' and 3' untranslated regions of mRNA).
Protein-coding sequences (specifically, coding exons) constitute less than 1.5% of the human genome.[3] In addition, about 26% of the human genome is introns.[8] Aside from genes (exons and introns) and known regulatory sequences (8–20%), the human genome contains regions of noncoding DNA. The exact amount of noncoding DNA that plays a role in cell physiology has been hotly debated. Recent analysis by the ENCODE project indicates that 80% of the entire human genome is either transcribed, binds to regulatory proteins, or is associated with some other biochemical activity.[2] It however remains controversial whether all of this biochemical activity contributes to cell physiology, or whether a substantial portion of this is the result transcriptional and biochemical noise, which must be actively filtered out by the organism.[9] Excluding protein-coding sequences, introns, and regulatory regions, much of the non-coding DNA is composed of: Many DNA sequences that do not play a role in gene expression have important biological functions. Comparative genomics studies indicate that about 5% of the genome contains sequences of noncoding DNA that are highly conserved, sometimes on time-scales representing hundreds of millions of years, implying that these noncoding regions are under strong evolutionary pressure and positive selection.[10] Many of these sequences regulate the structure of chromosomes by limiting the regions of heterochromatin formation and regulating structural features of the chromosomes, such as the telomeres and centromeres. Other noncoding regions serve as origins of DNA replication. Finally several regions are transcribed into functional noncoding RNA that regulate the expression of protein-coding genes (for example [11]), mRNA translation and stability (see miRNA), chromatin structure (including histone modifications, for example [12]), DNA methylation (for example [13]), DNA recombination (for example [14]), and cross-regulate other noncoding RNAs (for example [15]). It is also likely that many transcribed noncoding regions do not serve any role and that this transcription is the product of non-specific RNA Polymerase activity.[9]
Pseudogenes[]
Pseudogenes are inactive copies of protein-coding genes, often generated by gene duplication, that have become nonfunctional through the accumulation of inactivating mutations. Table 1 shows that the number of pseudogenes in the human genome is on the order of 13,000, and in some chromosomes is nearly the same as the number of functional protein-coding genes. Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution.
For example, the olfactory receptor gene family is one of the best-documented examples of pseudogenes in the human genome. More than 60 percent of the genes in this family are non-functional pseudogenes in humans. By comparison, only 20 percent of genes in the mouse olfactory receptor gene family are pseudogenes. Research suggests that this is a species-specific characteristic, as the most closely related primates all have proportionally fewer pseudogenes. This genetic discovery helps to explain the less acute sense of smell in humans relative to other mammals.[16]
Genes for noncoding RNA (ncRNA)[]
Noncoding RNA molecules play many essential roles in cells, especially in the many reactions of protein synthesis and RNA processing. The human genome contains genes encoding 18,400 ncRNAs, including tRNA, ribosomal RNA, microRNA, and other non-coding RNA genes.[2][17]
One historical misconception regarding the ncRNAs is that they lack critical genetic information or function. Rather, these ncRNAs are often critical elements in gene regulation and expression. Noncoding RNA also contributes to epigenetics, transcription, RNA splicing, and the translational machinery. The role of RNA in genetic regulation and disease offers a new potential level of unexplored genomic complexity.[18]
Introns and untranslated regions of mRNA[]
In addition to the ncRNA molecules that are encoded by discrete genes, the initial transcripts of protein coding genes usually contain extensive noncoding sequences, in the form of introns, 5'-untranslated regions (5'-UTR), and 3'-untranslated regions (3'-UTR). Within most protein-coding genes of the human genome, the length of intron sequences is 10- to 100-times the length of exon sequences (Table 2).
Regulatory DNA sequences[]
The human genome has many different regulatory sequences which are crucial to controlling gene expression. Known examples of such sequences make up 8% of the genome,[19] and extrapolating from ENCODE data give that 20[20]-40%[21] of the genome is gene regulatory sequence. Some types of non-coding DNA are genetic "switches" that do not encode proteins, but do regulate when and where genes are expressed (called enhancers).[22]
Regulatory sequences have been known since the late 1960s.[23] The first identification of regulatory sequences in the human genome relied on recombinant DNA technology.[24] Later with the advent of genomic sequencing, the identification of these sequences could be inferred by evolutionary conservation. The evolutionary branch between the primates and mouse, for example, occurred 70–90 million years ago.[25] So computer comparisons of gene sequences that identify conserved non-coding sequences will be an indication of their importance in duties such as gene regulation.[26] Other genomes have been sequenced with the same intention of aiding conservation-guided methods, for exampled the pufferfish genome.[27] However, regulatory sequences disappear and re-evolve during evolution at a high rate.[28][29][30] As of 2012, the efforts have shifted toward finding interactions between DNA and regulatory proteins by the technique ChIP-Seq, or gaps where the DNA is not packaged by histones (DNase hypersensitive sites), both of which tell where there are active regulatory sequences in the investigated cell type.[19]
Repetitive DNA sequences[]
About 8% of the human genome consists of repetitive DNA sequences, termed tandem DNA arrays or tandem repeats. The repeated sequences may be of variable lengths, from two nucleotides to tens of nucleotides. These sequences are highly variable, even among closely related individuals, and so are used for genealogical DNA testing and forensic DNA analysis.
Repeated sequences of fewer then ten nucleotides (e.g. the dinucleotide repeat (AC)n) are termed microsatellite sequences. Among the microsatellite sequences, trinucleotide repeats are of particular importance, as sometimes occur within coding regions of genes for proteins and may lead to genetic disorders. For example, Huntington's disease results from an expansion of the trinucleotide repeat (CAG)n within the Huntingtin gene on human chromosome 4. Telomeres (the ends of linear chromosomes) end with a microsatellite hexanucleotide repeat of the sequence (TTAGGG)n.
Tandem repeats of longer sequences (arrays of repeated sequences 10–60 nucleotides long) are termed minisatellites.
Mobile genetic elements (transposons) and their relics[]
Transposable genetic elements, DNA sequences that can replicate and insert copies of themselves at other locations within a host genome, are an abundant component in the human genome. The most abundant transposon lineage, Alu, has about 50,000 active copies,[31] while another lineage, LINE-1, has about 100 active copies per genome (the number varies between people).[32] Together with non-functional relics of old transposons, they account for over half of total human DNA.[33] Sometimes called "jumping genes", transposons have played a major role in sculpting the human genome. Some of these sequences represent endogenous retroviruses, DNA copies of viral sequences that have become permanently integrated into the genome and are now passed on to succeeding generations.
Mobile elements within the human genome can be classified into LTR retrotransposons (8.3% of total genome), SINEs (13.1% of total genome) including Alu elements, LINEs (20.4% of total genome), and Class II DNA transposons (2.9% of total genome).
Genomic variation in humans[]
The Human Reference Genome[]
With the exception of identical twins, all humans show significant variation in genomic DNA sequences. The Human Reference Genome (HRG) is used as a standard sequence reference.
There are several important points concerning the Human Reference Genome--
- The HRG is a haploid sequence. Each chromosome is represented once.
- The HRG is a composite sequence, and does not correspond to any actual human individual.
- The HRG is periodically updated to correct errors and ambiguities.
- The HRG in no way represents an "ideal" or "perfect" human individual. It is simply a standardized representation or model that is used for comparative purposes.
Measuring human genetic variation[]
Most studies of human genetic variation have focused on single-nucleotide polymorphisms (SNPs), which are substitutions in individual bases along a chromosome. Most analyses estimate that SNPs occur 1 in 1000 base pairs, on average, in the euchromatic human genome, although they do not occur at a uniform density. Thus follows the popular statement that "we are all, regardless of race, genetically 99.9% the same",[34] although this would be somewhat qualified by most geneticists. For example, a much larger fraction of the genome is now thought to be involved in copy number variation.[35] A large-scale collaborative effort to catalog SNP variations in the human genome is being undertaken by the International HapMap Project.
The genomic loci and length of certain types of small repetitive sequences are highly variable from person to person, which is the basis of DNA fingerprinting and DNA paternity testing technologies. The heterochromatic portions of the human genome, which total several hundred million base pairs, are also thought to be quite variable within the human population (they are so repetitive and so long that they cannot be accurately sequenced with current technology). These regions contain few genes, and it is unclear whether any significant phenotypic effect results from typical variation in repeats or heterochromatin.
Most gross genomic mutations in gamete germ cells probably result in inviable embryos; however, a number of human diseases are related to large-scale genomic abnormalities. Down syndrome, Turner Syndrome, and a number of other diseases result from nondisjunction of entire chromosomes. Cancer cells frequently have aneuploidy of chromosomes and chromosome arms, although a cause and effect relationship between aneuploidy and cancer has not been established.
Mapping human genomic variation[]
Whereas a genome sequence lists the order of every DNA base in a genome, a genome map identifies the landmarks. A genome map is less detailed than a genome sequence and aids in navigating around the genome.[36][37]
An example of a variation map is the HapMap being developed by the International HapMap Project. The HapMap is a haplotype map of the human genome, "which will describe the common patterns of human DNA sequence variation."[38] It catalogs the patterns of small-scale variations in the genome that involve single DNA letters, or bases.
Researchers published the first sequence-based map of large-scale structural variation across the human genome in the journal Nature in May 2008.[39][40] Large-scale structural variations are differences in the genome among people that range from a few thousand to a few million DNA bases; some are gains or losses of stretches of genome sequence and others appear as re-arrangements of stretches of sequence. These variations include differences in the number of copies individuals have of a particular gene, deletions, translocations and inversions.
Personal genomes[]
A personal genome sequence is a (nearly) complete sequence of the chemical base pairs that make up the DNA of a single person. Because medical treatments have different effects on different people because of genetic variations such as single-nucleotide polymorphisms (SNPs), the analysis of personal genomes may lead to personalized medical treatment based on individual genotypes.
The first personal genome sequence to be determined was that of Craig Venter in 2007. Personal genomes had not been sequenced in the public Human Genome Project to protect the identity of volunteers who provided DNA samples. That sequence was derived from the DNA of several volunteers from a diverse population.[41] However, early in the Venter-led Celera Genomics genome sequencing effort the decision was made to switch from sequencing a composite sample to using DNA from a single individual, later revealed to have been Venter himself. Thus the Celera human genome sequence released in 2000 was largely that of one man. Subsequent replacement of the early composite-derived data and determination of the diploid sequence, representing both sets of chromosomes, rather than a haploid sequence originally reported, allowed the release of the first personal genome.[42] In April 2008, that of James Watson was also completed. Since then hundreds of personal genome sequences have been released,[43] including those of Desmund Tutu,[44][45] and of a Paleo-Eskimo.[46]
The sequencing of individual genomes further unveiled levels of genetic complexity that had not been appreciated before. Personal genomics helped reveal the significant level of diversity in the human genome attributed not only to SNPs but structural variations as well. However, the application of such knowledge to the treatment of disease and in the medical field is only in its very beginnings.[47] Exome sequencing has become increasingly popular as a tool to aid in diagnosis of genetic disease because the exome comprises only 1% of the genomic sequence but accounts for roughly 85% of the disease causing mutations.[48]
Human genetic disorders[]
- For more details on this topic, see Genetic disorder.
Most aspects of human biology involve both genetic (inherited) and non-genetic (environmental) factors. Some inherited variation influences aspects of our biology that are not medical in nature (height, eye color, ability to taste or smell certain compounds, etc.). Moreover, some genetic disorders only cause disease in combination with the appropriate environmental factors (such as diet). With these caveats, genetic disorders may be described as clinically defined diseases caused by genomic DNA sequence variation. In the most straightforward cases, the disorder can be associated with variation in a single gene. For example, cystic fibrosis is caused by mutations in the CFTR gene, and is the most common recessive disorder in caucasian populations with over 1,300 different mutations known. Disease-causing mutations in specific genes are usually severe in terms of gene function, and are fortunately rare, thus genetic disorders are similarly individually rare. However, since there are many genes that can vary to cause genetic disorders, in aggregate they comprise a significant component of known medical conditions, especially in pediatric medicine. Molecularly characterized genetic disorders are those for which the underlying causal gene has been identified, currently there are approximately 2,200 such disorders annotated in the OMIM database.[49]
Studies of genetic disorders are often performed by means of family-based studies. In some instances population based approaches are employed, particularly in the case of so-called founder populations such as those in Finland, French-Canada, Utah, Sardinia, etc. Diagnosis and treatment of genetic disorders are usually performed by a geneticist-physician trained in clinical/medical genetics. The results of the Human Genome Project are likely to provide increased availability of genetic testing for gene-related disorders, and eventually improved treatment. Parents can be screened for hereditary conditions and counselled on the consequences, the probability it will be inherited, and how to avoid or ameliorate it in their offspring.
As noted above, there are many different kinds of DNA sequence variation, ranging from complete extra or missing chromosomes down to single nucleotide changes. It is generally presumed that much naturally occurring genetic variation in human populations is phenotypically neutral, i.e. has little or no detectable effect on the physiology of the individual (although there may be fractional differences in fitness defined over evolutionary time frames). Genetic disorders can be caused by any or all known types of sequence variation. To molecularly characterize a new genetic disorder, it is necessary to establish a causal link between a particular genomic sequence variant and the clinical disease under investigation. Such studies constitute the realm of human molecular genetics.
With the advent of the Human Genome and International HapMap Project, it has become feasible to explore subtle genetic influences on many common disease conditions such as diabetes, asthma, migraine, schizophrenia, etc. Although some causal links have been made between genomic sequence variants in particular genes and some of these diseases, often with much publicity in the general media, these are usually not considered to be genetic disorders per se as their causes are complex, involving many different genetic and environmental factors. Thus there may be disagreement in particular cases whether a specific medical condition should be termed a genetic disorder. The categorized table below provides the prevalence as well as the genes or chromosomes associated with some human genetic disorders.
| Disorder | Prevalence | Chromosome or Gene Involved |
|---|---|---|
| Chromosomal Conditions | ||
| Down Syndrome | 1:600 | Chromosome 21 |
| Klinefelter Syndrome | 1:500–1000 males | Additional X Chromosome |
| Turner Syndrome | 1:2000 females | Loss of X Chromsome |
| Cancers | ||
| Breast/Ovarian Cancer (susceptibility) | ~5% of cases of these cancer types | BRCA1, BRCA2 |
| FAP (hereditary nonpolyposis coli) | 1:3500 | APC |
| Lynch syndrome | 5–10% of all cases of bowel cancer | MLH1, MSH2, MSH6, PMS2 |
| Neurological Conditions | ||
| Huntington disease | 1:20000 | Huntingtin |
| Alzheimer disease ‐ early onset | 1:2500 | PS1, PS2, APP |
| Other Conditions | ||
| Cystic fibrosis | 1:2500 | CFTR |
| Muscular dystrophy – Duchenne type | 1:3500 boys | Dystrophin |
Evolution[]
Comparative genomics studies of mammalian genomes suggest that approximately 5% of the human genome has been conserved by evolution since the divergence of extant lineages approximately 200 million years ago, containing the vast majority of genes.[50][51] The published chimpanzee genome differs from that of the human genome by 1.23% in direct sequence comparisons.[52] Around 20% of this figure is accounted for by variation within each species, leaving only ~1.06% consistent sequence divergence between humans and chimps at shared genes.[53] This nucleotide by nucleotide difference is dwarfed, however, by the portion of each genome that is not shared, including around 6% of functional genes that are unique to either humans or chimps.[54] In other words, the considerable observable differences between humans and chimps may be due as much or more to genome level variation in the number, function and expression of genes rather than DNA sequence changes in shared genes. Indeed, even within humans, there has been found to be a previously unappreciated amount of copy number variation (CNV) which can make up as much as 5 – 15% of the human genome. In other words, between humans, there could be +/- 500,000,000 base pairs of DNA, some being active genes, others inactivated, or active at different levels. The full significance of this finding remains to be seen. On average, a typical human protein-coding gene differs from its chimpanzee ortholog by only two amino acid substitutions; nearly one third of human genes have exactly the same protein translation as their chimpanzee orthologs. A major difference between the two genomes is human chromosome 2, which is equivalent to a fusion product of chimpanzee chromosomes 12 and 13[55] (later renamed to chromosomes 2A and 2B, respectively).
Humans have undergone an extraordinary loss of olfactory receptor genes during our recent evolution, which explains our relatively crude sense of smell compared to most other mammals. Evolutionary evidence suggests that the emergence of color vision in humans and several other primate species has diminished the need for the sense of smell.[56]
Mitochondrial DNA[]
The human mitochondrial DNA is of tremendous interest to geneticists, since it undoubtedly plays a role in mitochondrial disease. It also sheds light on human evolution; for example, analysis of variation in the human mitochondrial genome has led to the postulation of a recent common ancestor for all humans on the maternal line of descent. (see Mitochondrial Eve)
Due to the lack of a system for checking for copying errors, Mitochondrial DNA (mtDNA) has a more rapid rate of variation than nuclear DNA. This 20-fold increase in the mutation rate allows mtDNA to be used for more accurate tracing of maternal ancestry. Studies of mtDNA in populations have allowed ancient migration paths to be traced, such as the migration of Native Americans from Siberia or Polynesians from southeastern Asia. It has also been used to show that there is no trace of Neanderthal DNA in the European gene mixture inherited through purely maternal lineage.[57]
Epigenome[]
Epigenetics describes a variety of features of the human genome that transcend its primary DNA sequence, such as chromatin packaging, histone modifications and DNA methylation, and which are important in regulating gene expression, genome replication and other cellular processes. Epigenetic markers strengthen and weaken transcription of certain genes but do not affect the actual sequence of DNA nucleotides. DNA methylation is a major form of epigenetic control over gene expression and one of the most highly studied topics in epigenetics. During development, the human DNA methylation profile experiences dramatic changes. In early germ line cells, the genome has very low methylation levels. These low levels generally describe active genes. As development progresses, parental imprinting tags lead to increased methylation activity.[58][59]
Epigenetic patterns can be identified between tissues within an individual as well as between individuals themselves. Identical genes that have differences only in their epigenetic state are called epialleles. Epialleles can be placed into three categories: those directly determined by an individual’s genotype, those influenced by genotype, and those entirely independent of genotype. The epigenome is also influenced significantly by environmental factors. Diet, toxins, and hormones impact the epigenetic state. Studies in dietary manipulation have demonstrated that methyl-deficient diets are associated with hypomethylation of the epigenome. Such studies establish epigenetics as an important interface between the environment and the genome.[60]
See also[]
- Craig Venter#Individual human genome sequenced
- Eugenics
- Eukaryotic chromosome fine structure
- Genetic distance
- Genomic organization
- Human genetic engineering
- Human Genome Sequencing Center
- Full genome sequencing
- Pharmacogenetics
- Noncoding DNA
- The Genographic Project
- Mitochondrial Eve
- Y-chromosomal Adam
References[]
- ^ International Human Genome Sequencing Consortium (2004). "Finishing the euchromatic sequence of the human genome.". Nature 431 (7011): 931–45. DOI:10.1038/nature03001. PMID 15496913. [1]
- ^ a b c d Elizabeth Pennisi (2012). "ENCODE Project Writes Eulogy For Junk DNA". Science 337 (6099): 1159–1160. DOI:10.1126/science.337.6099.1159.
- ^ a b c International Human Genome Sequencing Consortium (2001). "Initial sequencing and analysis of the human genome". Nature 409 (6822): 860–921. DOI:10.1038/35057062. PMID 11237011. [2] Cite error: Invalid
<ref>tag; name "IHSGC2001" defined multiple times with different content - ^ PANTHER Pie Chart at the PANTHER Classification System homepage. Retrieved May 25, 2011
- ^ (1969) "Metabolic stability and epigenesis in randomly constructed genetic nets". Journal of Theoretical Biology 22 (3): 437–467. DOI:10.1016/0022-5193(69)90015-0. PMID 5803332.
- ^ (1972) "An argument for the genetic simplicity of man and other mammals". Journal of Human Evolution 1 (6): 651–662. DOI:10.1016/0047-2484(72)90011-5.
- ^ M. Huang, H. Zhu, B. Shen, G. Gao, "A non-random gait through the human genome", 3rd International Conference on Bioinformatics and Biomedical Engineering (UCBBE, 2009), 1–3
- ^ (2005) "Synergy between sequence and size in Large-scale genomics". Nature Reviews Genetics 6 (9): 699–708. DOI:10.1038/nrg1674. ISSN 1471-0056.
- ^ a b (2012) "Nuclear export as a key arbiter of "mRNA identity" in eukaryotes". Biochimica et Biophysica Acta (BBA) – Gene Regulatory Mechanisms 1819 (6): 566–577. DOI:10.1016/j.bbagrm.2011.12.012. ISSN 18749399.
- ^ Ludwig MZ (December 2002). "Functional evolution of noncoding DNA". Curr. Opin. Genet. Dev. 12 (6): 634–9. DOI:10.1016/S0959-437X(02)00355-6. PMID 12433575.
- ^ (2004) "Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene". Nature 429 (6991): 571–574. DOI:10.1038/nature02538. ISSN 0028-0836.
- ^ (2010) "Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes". Science 329 (5992): 689–693. DOI:10.1126/science.1192002. ISSN 0036-8075.
- ^ (1991) "Parental imprinting of the mouse H19 gene". Nature 351 (6322): 153–155. DOI:10.1038/351153a0. ISSN 0028-0836.
- ^ (2005) "Recombination Regulation by Transcription-Induced Cohesin Dissociation in rDNA Repeats". Science 309 (5740): 1581–1584. DOI:10.1126/science.1116102. ISSN 0036-8075.
- ^ (2011) "A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language?". Cell 146 (3): 353–358. DOI:10.1016/j.cell.2011.07.014. ISSN 00928674.
- ^ Gilad, Y; Man, O; Pääbo, S; Lancet, D (2003 Mar 18). "Human specific loss of olfactory receptor genes.". Proceedings of the National Academy of Sciences of the United States of America 100 (6): 3324–7. PMID 12612342.
- ^ (2001) "Non-coding RNA genes and the modern RNA world". Nature Reviews Genetics 2 (12): 919–929. DOI:10.1038/35103511. PMID 11733745.
- ^ Mattick, JS; Makunin, IV (2006 Apr 15). "Non-coding RNA.". Human molecular genetics 15 Spec No 1: R17-29. PMID 16651366.
- ^ a b doi:10.1038/nature11247
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ Ewan Birney, http://genomeinformatician.blogspot.ca/2012/09/encode-my-own-thoughts.html
- ^ doi:10.1101/gr.146506.112
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ Carroll, Sean B. et al. (May 2008). "Regulating Evolution", Scientific American, pp. 60–67.
- ^ (1968) "The promoter-operator region of the Lac operon of Escherichia coli". Journal of Molecular Biology 38 (3): 413–420. DOI:10.1016/0022-2836(68)90395-1. ISSN 00222836.
- ^ (1984) "DNA sequences required for regulated expression of β-globin genes in murine erythroleukemia cells". Cell 38 (1): 265–273. DOI:10.1016/0092-8674(84)90548-8. ISSN 00928674.
- ^ Nei M, Xu P, Glazko G (2001). "Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms". Proc Natl Acad Sci USA 98 (5): 2497–502. DOI:10.1073/pnas.051611498. PMID 11226267.
- ^ Loots G, Locksley R, Blankespoor C, Wang Z, Miller W, Rubin E, Frazer K (2000). "Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons". Science 288 (5463): 136–40. DOI:10.1126/science.288.5463.136. PMID 10753117. Summary
- ^ Meunier, Monique. "Genoscope and Whitehead announce a high sequence coverage of the Tetraodon nigroviridis genome". Genoscope. Archived from the original on 16 October 2006. http://web.archive.org/web/20061016085223/http://www.cns.fr/externe/English/Actualites/Presse/261001_1.html. Retrieved 2006-09-12.
- ^ doi:10.1038/nrg3229
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ doi:10.1126/science.1186176
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ doi:10.1126/science.1160930
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ doi:10.1101/gr.081737.108
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ doi:10.1073/pnas.0831042100
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ^ Nicholas H. Barton, Derek E. G. Briggs, Jonathan A. Eisen, David B. Goldstein, Nipam H. Patel "Evolution" Cold Spring Harbor Laboratory Press; 1st edition (June 30, 2007) ISBN 0-87969-684-2
- ^ from Bill Clinton's 2000 State of the Union address
- ^ Nature. "Global variation in copy number in the human genome : Article : Nature". Nature. http://www.nature.com/nature/journal/v444/n7118/full/nature05329.html. Retrieved 2009-08-09.
- ^ "What's a Genome?". Genomenewsnetwork.org. 2003-01-15. http://www.genomenewsnetwork.org/resources/whats_a_genome/Chp3_1.shtml. Retrieved 2009-05-31.
- ^ NCBI_user_services (2004-03-29). "Mapping Factsheet". Ncbi.nlm.nih.gov. http://www.ncbi.nlm.nih.gov/About/primer/mapping.html. Retrieved 2009-05-31.
- ^ "About the Project". HapMap. http://www.hapmap.org/abouthapmap.html. Retrieved 2009-05-31.
- ^ "2008 Release: Researchers Produce First Sequence Map of Large-Scale Structural Variation in the Human Genome". genome.gov. http://www.genome.gov/27026113. Retrieved 2009-05-31.
- ^ Nature. "Mapping and sequencing of structural variation from eight human genomes : Abstract". Nature. doi:10.1038/nature06862. http://www.nature.com/nature/journal/v453/n7191/abs/nature06862.html. Retrieved 2009-05-31.
- ^ "Human Genome Project Completion: Frequently Asked Questions". genome.gov. http://www.genome.gov/11006943. Retrieved 2009-05-31.
- ^ Singer, Emily (September 4, 2007). "Technology Review". Technology review. http://www.technologyreview.com/biomedicine/19328/?a=f. Retrieved May 25, 2010.
- ^ http://www.completegenomics.com/news-events/press-releases/Complete-Genomics-Adds-29-High-Coverage-Complete-Human-Genome-Sequencing-Datasets-to-its-Public-Genomic-Repository--119298369.html
- ^ [3] Desmond Tutu's genome sequenced as part of genetic diversity study], Ian Sample, The Guardian, 17 February 2010.
- ^ Complete Khoisan and Bantu genomes from southern Africa; Schuster, Stephan C. Nature 463, 943–947 (18 February 2010)
- ^ (Feb 2010) "Ancient human genome sequence of an extinct Palaeo-Eskimo.". Nature 463 (7282): 757–62. DOI:10.1038/nature08835.
- ^ Gonzaga-Jauregui, C; Lupski, JR; Gibbs, RA (2012). "Human genome sequencing in health and disease.". Annual review of medicine 63: 35–61. PMID 22248320.
- ^ Choi, M.; Scholl, U. I.; Ji, W.; Liu, T.; Tikhonova, I. R.; Zumbo, P.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Sanjad, S.; Nelson-Williams, C.; Farhi, A.; Mane, S.; Lifton, R. P. (27 October 2009). "Genetic diagnosis by whole exome capture and massively parallel DNA sequencing". Proceedings of the National Academy of Sciences 106 (45): 19096–19101. DOI:10.1073/pnas.0910672106.
- ^ Online Mendelian Inheritance in Man (OMIM)
- ^ "...the proportion of small (50–100 bp) segments in the mammalian genome that is under (purifying) selection can be estimated to be about 5%. This proportion is much higher than can be explained by protein-coding sequences alone, implying that the genome contains many additional features (such as untranslated regions, regulatory elements, non-protein-coding genes, and chromosomal structural elements) under selection for biological function." Mouse Genome Sequencing Consortium (2002). "Initial sequencing and comparative analysis of the mouse genome". Nature 420 (6915): 520–62. DOI:10.1038/nature01262. PMID 12466850.
- ^ The ENCODE Project Consortium (2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature 447 (7146): 799–816. DOI:10.1038/nature05874. PMID 17571346.
- ^ "We calculate the genome-wide nucleotide divergence between human and chimpanzee to be 1.23%, confirming recent results from more limited studies. The Chimpanzee Sequencing and Analysis Consortium (2005). "Initial sequence of the chimpanzee genome and comparison with the human genome". Nature 437 (7055): 69–87. DOI:10.1038/nature04072. PMID 16136131.
- ^ "we estimate that polymorphism accounts for 14–22% of the observed divergence rate and thus that the fixed divergence is ~1.06% or less. The Chimpanzee Sequencing and Analysis Consortium (2005). "Initial sequence of the chimpanzee genome and comparison with the human genome". Nature 437 (7055): 69–87. DOI:10.1038/nature04072. PMID 16136131.
- ^ "Our results imply that humans and chimpanzees differ by at least 6% (1,418 of 22,000 genes) in their complement of genes, which stands in stark contrast to the oft-cited 1.5% difference between orthologous nucleotide sequences." Demuth J, De Bie T, Stajich JE, Cristianini N, Hahn MW (2006). "The Evolution of Mammalian Gene Families". PLoS ONE 1 (1): E85. DOI:10.1371/journal.pone.0000085. PMID 17183716.
- ^ "Human chromosome 2 resulted from a fusion of two ancestral chromosomes that remained separate in the chimpanzee lineage" The Chimpanzee Sequencing and Analysis Consortium (2005). "Initial sequence of the chimpanzee genome and comparison with the human genome". Nature 437 (7055): 69–87. DOI:10.1038/nature04072. PMID 16136131.
"Large-scale sequencing of the chimpanzee genome is now imminent."Olson M, Varki A (2003). "Sequencing the chimpanzee genome: insights into human evolution and disease". Nat Rev Genet 4 (1): 20–8. DOI:10.1038/nrg981. PMID 12509750. - ^ "Our findings suggest that the deterioration of the olfactory repertoire occurred concomitant with the acquisition of full trichromatic color vision in primates." Gilad Y, Wiebe V, Przeworski M, Lancet D, Pääbo S (2004). "Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates". PLoS Biol 2 (1). DOI:10.1371/journal.pbio.0020005. PMID 14737185.
- ^ Sykes, Bryan (2003-10-09). "Mitochondrial DNA and human history". The Human Genome. http://genome.wellcome.ac.uk/doc_WTD020876.html. Retrieved 2006-09-19.
- ^ "Misteli". Cell. doi:10.1016/j.cell.2007.01.028. http://www.cell.com/content/article/abstract?uid=PIIS0092867407001262. Retrieved 2009-05-31.
- ^ "Bernstein et al.". Cell. doi:10.1016/j.cell.2007.01.033. http://www.cell.com/content/article/abstract?uid=PIIS0092867407001286. Retrieved 2009-05-31.
- ^ Scheen, AJ; Junien, C (2012 May–Jun). "[Epigenetics, interface between environment and genes: role in complex diseases].". Revue medicale de Liege 67 (5–6): 250–7. PMID 22891475.
- Carroll, Sean B. et al. (May 2008). "Regulating Evolution". Scientific American. New York: Scientific American Inc.. pp. 60–67.
- Lindblad-Toh K, et al. (2005). "Genome sequence, comparative analysis and haplotype structure of the domestic dog". Nature 438 (7069): 803–19. DOI:10.1038/nature04338. PMID 16341006.
External links[]
| Wikinews has related news: Mexico presents first population-wide genome map for a Latin country |
- The National Human Genome Research Institute
- Ensembl The Ensembl Genome Browser Project
- National Library of Medicine human genome viewer
- UCSC Genome Browser.
- Human Genome Project.
- The National Office of Public Health Genomics
- New findings challenge established views about human genome
- INMEGEN: Complete genetic map of some mexican native groups
- Missing bits of DNA may define humans
| |||||
| ||||||||
| This page uses content from the English language Wikipedia. The original content was at Human genome. The list of authors can be seen in the page history. As with this Familypedia wiki, the content of Wikipedia is available under the Creative Commons License. |